
In pharmaceutical development, the pharmacokinetic (PK) statistical review serves as a crucial link between biological data and regulatory acceptance, establishing a mathematical basis for a drug’s expected behavior in the human body. Key PK parameters such as Cmax, AUC, Tmax, and half-life form the foundation of drug applications and approvals. However, these parameters gain significance only when supported by a robust statistical framework that assures regulators of the drug’s safety and effectiveness. Regulators are interested not just in the PK numbers, but in the statistical confidence that validates the conclusions drawn from these measurements.
This is an overview of PK-Stat Review, which employs statistical methods to analyze pharmacokinetic data, aiming to derive dependable insights regarding drug behavior, bioequivalence, and therapeutic efficacy. The thoroughness of this review is crucial for sponsors in the regulatory approval process, as it can significantly influence the progression of an application—potentially allowing it to succeed rapidly or hindering it indefinitely.
👉 Transform PK Data into Regulatory Confidence
Zenovel helps sponsors convert complex pharmacokinetic datasets into submission-ready statistical evidence aligned with global regulatory expectations.
Zenovel’s PK-Stat practice leverages with years of regulatory expertise, serving varied important clients globally. The blog highlights the importance of PK-Stat review in bioequivalence studies and regulatory submissions, emphasizing how Zenovel ensures that complex pharmacokinetic data is converted into persuasive regulatory evidence.
What Is PK-Stat Review?
PK-Stat Review involves the statistical analysis and interpretation of pharmacokinetic data from clinical studies, covering the process from raw concentration-time data to final regulatory outcomes.
The scope of PK-Stat Review includes:

Figure 1: Zenovel’s PK Stat Review Methods
The final objective is to provide regulators with irrefutable evidence that the drug product performs as intended, with known and acceptable variability.
Why PK-Stat Review Matters?
Regulatory agencies such as the US FDA, EMA, MHRA, and CDSCO demand thorough statistical proof of bioequivalence and pharmacokinetic performance, with serious consequences for insufficient analysis.
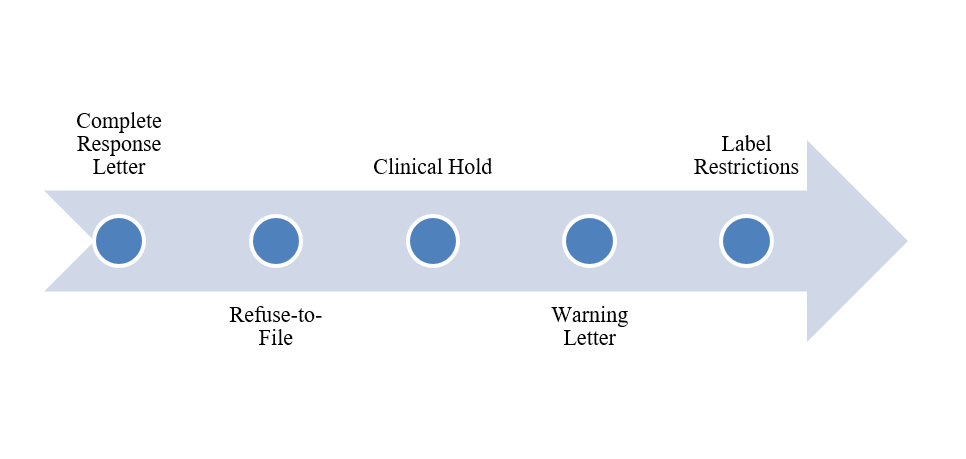
Figure 2: Consequences of non-compliance
| Consequences | It Effect |
| Complete Response Letter | Approval has been delayed by 6 to 12 months due to the need to address deficiencies.
|
| Refuse-to-File | Submission was rejected prior to the commencement of the review process.
|
| Clinical Hold | Ongoing trials have been suspended while awaiting data reanalysis.
|
| Warning Letter | Public disclosure of compliance failures |
| Label Restrictions | Narrower indications or dosing limitations |
FDA guidance on bioequivalence studies outlines specific statistical requirements for sponsors. Non-compliance results in disapproval.
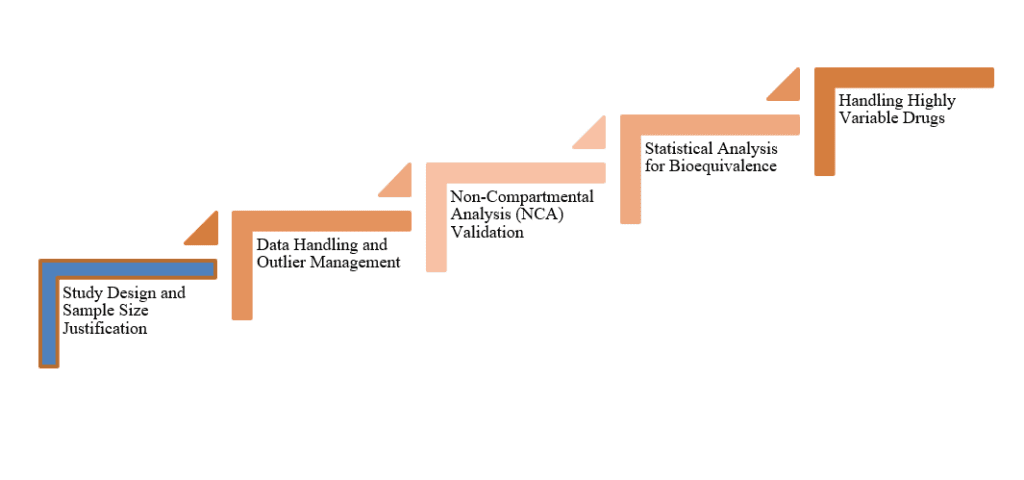
Core Components of PK-Stat Review in Bioequivalence Studies
 Study Design and Sample Size Justification
Study Design and Sample Size Justification
 Study Design and Sample Size Justification
Study Design and Sample Size JustificationBefore administering a dose, a statistical foundation is essential, starting with sample size calculation for demonstrating bioequivalence with appropriate power. Key considerations include expected intra-subject variability for Cmax and AUC, desired power (typically 80-90%), significance level (α = 0.05 for two one-sided tests), expected geometric mean ratio (T/R), and the study design (crossover, parallel, replicate). The modeling and simulation approach is increasingly important, as shown by a study that reduced required sample size from 40 to 20 subjects while maintaining over 90% power, ultimately saving time and costs.
Data Handling and Outlier Management
Raw concentration-time data necessitates meticulous preprocessing for effective statistical analysis. PK-Stat review focuses on handling missing data, outliers, partial profiles, and ensuring data integrity through ALCOA+ compliance.
Non-Compartmental Analysis (NCA) Validation
Executing NCA requires precision. The PK-Stat review ensures accurate AUC calculations via trapezoidal methods, limits AUC0-∞ extrapolation to under 20%, reliably estimates the terminal elimination rate constant (λz), and correctly determines Cmax and Tmax. Errors in this phase affect the entire analysis.
Statistical Analysis for Bioequivalence
The assessment of bioequivalence utilizes the two one-sided tests (TOST) procedure, focusing on the appropriateness of the ANOVA model, which considers factors such as sequence, period, formulation, and subject. It involves constructing 90% confidence intervals for the ratio of geometric means (T/R), which must fall within 80.00-125.00% for both Cmax and AUC to satisfy bioequivalence criteria. Additionally, estimating intra-subject variability is important, especially for replicate designs and highly variable drugs.
Handling Highly Variable Drugs
Some drugs demonstrate high intra-subject variability (CV% >30%), complicating traditional bioequivalence assessments. The Reference-Scaled Average Bioequivalence (RSABE) method addresses this by scaling bioequivalence limits according to the reference product’s within-subject variability. It necessitates replicate crossover designs (such as TRTR, RTRT, or partial designs like TRR, RTR, RRT) and requires enrollment of at least 24 subjects.
PK-Stat Review in Regulatory Submissions: CTD Module 2
In the Common Technical Document (CTD), PK-Stat Review findings are primarily presented in Module 2, specifically in Sections 2.7.1 and 2.7.2.
Section 2.7.1: Summary of Biopharmaceutics Studies and Associated Analytical Methods
This section summarizes bioavailability and bioequivalence studies and their associated analytical methods.
- Food effect studies
- Relative bioavailability comparisons between formulations
- In vitro dissolution data at different strengths and pH conditions
- Solubility and permeability assessments
- Bioanalytical method summaries include tables with information on method lifecycle, performance characteristics, and validation results. The FDA’s 2019 guidance mandates tabular summaries for all clinical studies, necessitating specialized expertise for accurate compilation.
Section 2.7.2: Summary of Clinical Pharmacology Studies
This section offers a comprehensive overview of all studies producing pharmacokinetic (PK) and pharmacodynamic (PD) data.
- Single and multiple ascending dose studies
- Mass balance and metabolite identification (ADME) studies
- Drug-drug interaction evaluations
- Organ impairment studies
👉 Simplify CTD Module 2 Authoring
Zenovel helps prepare structured, regulator-friendly PK and biopharmaceutics summaries for faster review readiness.
Population PK and exposure-response analyses
Immunogenicity data integration
Section 2.7.2 faces the challenge of encompassing extensive content, often spanning hundreds of pages and integrating numerous studies alongside thousands of pharmacokinetic observations, necessitating the translation of complex quantitative data into a coherent regulatory narrative.
Best practices for authoring :
- Start with key takeaways upfront
- Cross-reference other sections instead of repeating data
- Use relevant tables and figures strategically
- Ensure consistency with Module 2.5 (Clinical Overview)
- Write for review efficiency—clear headings, concise language, logical flow
Submission of Pharmacometric Data
The FDA has defined specific requirements for the submission of pharmacometric datasets and models.
- Datasets should be submitted in either SAS transport (.xpt) or comma-delimited (.csv) file formats.
- Files can be defined as either XML or PDF formats, which describe various variables
- Reviewer’s Guide detailing the necessary software, version specifications, dependencies, and the correct order for execution.
- Model codes outline key building steps, including base, covariate, final, and validation stages. Additionally, individual plots for representative subjects illustrate observed concentrations alongside individual and population predictions.
- Parameter names should be presented with units (e.g., CL/F in L/h) rather than using abstract notation like THETA(1). Additionally, covariate assessments should focus on their effect on exposure parameters, rather than solely on model parameters.
Common PK-Stat Review Deficiencies
Regulatory submissions frequently falter on PK-Stat issues. Understanding common pitfalls helps avoid them:
| Deficiency Area
|
Typical Finding |
| Sample Size | Inadequate power due to underestimated variability |
| AUC Extrapolation | Extrapolated area exceeding 20% of total AUC |
| Statistical Model | Incorrect ANOVA model |
| Confidence Intervals | 90% CI calculation errors |
| RSABE Application | Applied without meeting variability thresholds or protocol pre-specification |
| Missing Data | Inadequate handling of BLQ values or early withdrawals |
| Missing Data | Inadequate handling of BLQ values or early withdrawals |
| Bioanalytical Methods | Incomplete validation or missing cross-validation |
Each deficiency represents potential months of delay and significant additional cost.
Zenovel’s PK-Stat Review Services

Zenovel offers thorough PK-Stat review services throughout the drug development lifecycle, leveraging extensive regulatory expertise and a successful track record serving clients globally to ensure rigorous statistical and regulatory support for submissions.
Why Zenovel for PK-Stat Review?

The Future of PK-Stat Review
👉 Stay Ahead in the Evolving PK Landscape
Leverage advanced statistical methodologies and regulatory insights to strengthen future-ready pharmacokinetic submissions.
PK-Stat review is evolving rapidly. Key trends shaping the future include:
References:
- FDA Guidance: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA
- PubMed: A 2024 Update on US FDA Implementation of Partial Area Under the Curve Into Bioavailability and Bioequivalence Assessments
- PubMed: Advancements in Virtual Bioequivalence: A Systematic Review