
Data integrity has transitioned from a marginal concern to the foremost compliance risk in pharmaceutical manufacturing as of 2026. Previously minor lapses are now viewed by regulators as indicative of systemic failures, leading to increasingly severe consequences such as import alerts, product holds, damage to reputation, and potential criminal prosecution. Global regulators emphasize the critical message that the reliability of data directly correlates with the trustworthiness of medicines produced, marking a significant shift in regulatory focus and urgency.
The New Era of Data Integrity Enforcement
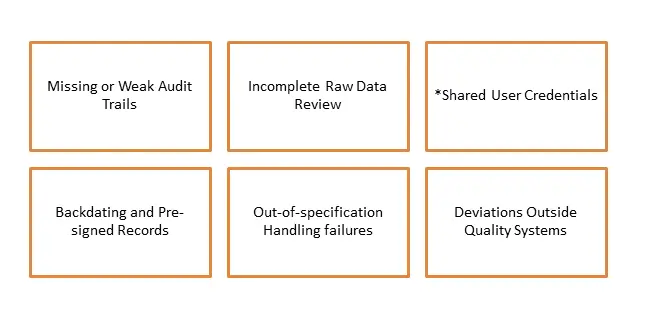
Regulatory enforcement by the FDA is significantly increasing in visibility in 2026, with FDA Warning Letters gaining renewed attention across various regulated industries. Unlike in previous years, the FDA is not only identifying isolated mistakes but is also searching for systemic issues. Specific concerns such as absent audit trails, backdated records, terminated or destroyed documents, and the use of shared logins are leading to intensified scrutiny. This rigorous examination can result in the freezing of product releases and the stalling of entire manufacturing operations.
👉 Is Your GMP Data Inspection-Ready?
Prevent audit trail gaps, documentation risks, and compliance failures before regulators find them.
Consider just a few examples from the past year:
In March 2026, the FDA addressed significant compliance failures at an Indian contract testing laboratory, as detailed in a Warning Letter. Investigators discovered two garbage bags filled with discarded analytical records, including chromatographic results and validation spreadsheets. The laboratory staff relied on unofficial personal diaries for recording critical procedures, results, and deviations, while official records only reflected handwritten values, lacking the original data. The laboratory contended that the discarded documents were irrelevant to products destined for the U.S. However, the FDA firmly stated that such documentation practices undermine data traceability and cast substantial doubt on the integrity of all data produced at the facility.
A US manufacturer in Washington state received a Warning Letter in February 2026 due to systemic quality control failures. Key issues identified included inadequate microbiological testing, absence of stability data, and insufficient oversight from the Quality Unit. These deficiencies significantly undermine confidence in the reliability of the laboratory data generated by the manufacturer.
Data integrity issues were mentioned in about 15% of 85 FDA Warning Letters issued to drug manufacturers in 2025, highlighting a significant and uneven distribution of these incidents.
- Indian sites received warning letters with data integrity findings at a staggering 60% rate
- China followed at 21%
- The United States at just 10%
This disparity suggests that data integrity challenges reflect deeper differences in quality culture across regional manufacturing landscapes.
Why Data Integrity Is the #1 Risk in 2026
- Data integrity failures lead to serious business repercussions for pharmaceutical companies, extending beyond just warning letters.
- Regulatory Fallout: Warning letters are publicly accessible and may result in various consequences, including delays in product approvals or submissions, import alerts, product holds, increased inspection frequency, and substantial reputational risks within the industry.
- Patient Safety Risks: When data is fabricated, deleted, or altered, the safety and efficacy of medicines are compromised. Incomplete or inconsistent batch records indicate manufacturing oversight gaps that affect patient care.
- Supply Chain Disruption: Data integrity red flags can lead to increased FDA scrutiny, longer remediation timelines, higher compliance costs, and negatively affect US sales if output or releases are delayed.
- Remediation Costs: The FDA recommended a GMP consultant in 87% of 2025 Warning Letters, highlighting that failures indicate broader systemic weaknesses that necessitate external expertise for remediation.
- Criminal Exposure: Severe data integrity violations can lead to criminal investigations if there is evidence of intent to deceive.
👉 Avoid Warning Letters Before They Happen
Zenovel helps pharmaceutical manufacturers strengthen GMP compliance, ALCOA+ controls, and audit readiness.
ALCOA+: The Universal Language of Data Integrity
ALCOA+ is a foundational framework for data integrity that regulators use to assess the trustworthiness of records. It requires that records be Attributable, Legible, Contemporaneous, Original, and Accurate. The “+” in ALCOA+ includes additional criteria: Complete, Consistent, Enduring, and Available.
div style=”border-left: 5px solid #f59221; padding: 20px; background: #fef9f4; margin: 30px 0; border-radius: 6px;”>
👉 Build a Strong ALCOA+ Framework
Implement secure audit trails, validated systems, and compliant documentation practices across your operations.
👉 Get ALCOA+ Compliance Support
In practice, this translates to non-negotiable requirements:
- Attributable: Unique user IDs, prohibition of shared accounts, e-signatures bound to identity.
- Contemporaneous: Real-time entries, no transcribing from uncontrolled “scratch pads”.
- Original: Retention of raw data and true copies with validated migration when digitized.
- Enduring/Available: Protected retention with reliable backup/restore and readable archives, rapidly retrievable for inspection.
Industry experts emphasize the importance of ALCOA+ for reliability, noting that it serves as a concise audit checklist. Regulators warn against attributing errors solely to “human error” without clarifying the exclusion of systemic or procedural factors. Operator mistakes might indicate deeper issues like unclear standard operating procedures, inadequate interface design, poor role separation, or lack of verification processes.
The Regulatory Landscape: What’s Changed in 2026
Both the FDA and EU regulators have raised their expectations regarding data integrity due to the growing complexity of digital systems and the essential nature of reliable data for pharmaceutical quality and patient safety.
FDA Focus Areas in 2025-2026
- Systemic Quality Culture: The FDA is transitioning its focus from singular procedural failures to overarching systemic issues, highlighting the significance of organizational culture in ensuring data integrity.
- Supplier and CMO Oversight: Increased scrutiny is being placed on companies concerning their management of contract manufacturers and suppliers, particularly related to data traceability and audit trails.
- Audit Trails and Metadata: The FDA mandates the preservation and accessibility of complete, secure, and reviewable audit trails, including metadata such as timestamps and user IDs.
- Remote Regulatory Assessments (RRAs): RRAs are permanent tools that require companies to keep data systems in an inspection-ready state continuously.
- AI and Predictive Oversight: The FDA has introduced AI tools to enhance the identification of high-risk inspection targets, emphasizing the importance of data transparency and integrity.
- Resilient Data Systems: Emphasizes data governance focusing on accuracy, ownership, and lifecycle management, advocating for learning from failures rather than repeating them.
div style=”border-left: 5px solid #f59221; padding: 20px; background: #fef9f4; margin: 30px 0; border-radius: 6px;”>
👉 Prepare for AI-Driven FDA Inspections
Stay ahead of evolving FDA expectations with risk-based validation and data governance strategies.
👉 Book a GMP Compliance Consultation
EMA Revisions (Final Publication Expected in 2026)
The EMA has introduced major draft revisions that fundamentally reshape expectations:
- Chapter 4 (Documentation): Document length has increased from 9 to 17 pages, with approximately 50% being completely rewritten. It now defines a lifecycle approach for documentation that incorporates data governance and ICH Q9 risk assessment. Hybrid systems for electronic signatures are advised against, and adherence to ALCOA++ principles is mandatory rather than a best practice.
- Annex 11 (Computerised Systems): Expanded from 9 to 19 pages, the document now includes increased expectations for lifecycle management, cybersecurity, identity and access management, audit trails, and supplier oversight.
- Annex 22 (Artificial Intelligence): Introduces the first formal GMP framework for AI, emphasizing clear intended use, robust model validation, and mandatory human oversight.
- GMDP IWG 3-Year Work Plan (2025-2027): Planned updates focus on data integrity, aiming for the final publication of Chapter 4 and Annex 11 amendments by Q1 2026.
- Organizations should promptly update procedures, assess gaps, enhance validation strategies, and strengthen AI oversight to ensure compliance.
Where Data Integrity Fails Most Often
The Future: AI, Digital Twins, and Human Oversight
AI is transforming pharmaceutical manufacturing by utilizing unstructured data to improve safety, quality, and efficiency. However, this technological shift brings data integrity challenges, prompting the EMA’s Annex 22, which requires clear intended use, strong model validation, and human oversight for AI systems in GMP settings. Conversely, AI can also enhance data integrity by identifying error-prone areas and utilizing digital twins for centralized data aggregation, facilitating quicker product releases.
The importance of a human-in-the-loop model, where human decision-makers assess AI-generated data to determine the safety of a batch before release, despite machines’ superior data processing capabilities.
Building a Data Integrity Program for 2026 and Beyond
- To differentiate compliant organizations from those receiving warning letters, a strong, proactive data governance framework is essential. Key components include:
- Governance and Policy: Implement lifecycle controls, define roles for data stewardship, and ensure regular independent reviews.
- Access Control and User Management: Use unique credentials, conduct regular access reviews, and separate system administration from data entry.
- Audit Trail Controls: Enable system-generated audit trails for all critical data, validate backup procedures, and ensure procedural ownership.
- Risk-Based CSV Strategy: Align validation with system impact, focusing on areas affecting patient safety and data integrity, following upcoming FDA guidance on risk-based documentation.
- Training and Quality Culture: Foster a culture of quality and continuous learning regarding data integrity among personnel.
- Supplier and CMO Oversight: Ensure compliance and documentation practices among contract partners.
- Remote Regulatory Assessments (RRAs): Maintain inspection-ready data systems.
Independent Assessments. - Develop comprehensive remediation plans including independent evaluations of laboratory systems as required by the FDA.
Zenovel provides comprehensive GMP compliance and data integrity solutions, helping pharmaceutical manufacturers meet global regulatory standards (FDA, EMA, CDSCO) with robust ALCOA+ frameworks, audit trail management, and risk-based computerized system validation.
Final Call.
Data integrity has escalated from a back-office issue to a critical boardroom concern affecting regulatory compliance, market access, patient safety, and corporate sustainability. By 2026, both the EMA’s GMP revisions and the FDA’s AI-driven inspections will enable regulators to swiftly identify systemic failures, with heightened consequences for non-compliance. Pharmaceutical manufacturers must integrate data integrity throughout their quality systems, adhering strictly to ALCOA+ principles and fostering a culture where data integrity is viewed as a collective responsibility.
Because in 2026, if your data can’t be trusted, neither can you.