
Bioequivalence (BE) studies are crucial in generic drug development to confirm that affordable generics are as safe and effective as brand-name drugs. However, proving equivalence for complex generics is a notably more complicated process compared to standard oral tablets.
What are bioequivalence studies and its importance?
BE studies confirm that generic drugs release their active ingredients to the site of action at a rate and extent comparable to brand-name drugs, demonstrated through pharmacokinetic parameters such as AUC and Cmax, requiring a range of 80-125% relative to the reference. These studies enable regulators like the FDA to approve generics through abbreviated processes, reducing costs, fostering competition, and enhancing patient access to medications. Without established bioequivalence, generics cannot replace brand-name drugs, impacting affordability and availability.
Complex generics present more challenges compared to simple generics, which include immediate-release oral solid dosage forms like standard tablets or capsules. Simple generics can often depend on basic in vivo pharmacokinetic studies in healthy volunteers or biowaivers based on the Biopharmaceutics Classification System.
Complex generics present significant challenges due to their inherent characteristics, as indicated by the FDA.
active ingredients (e.g., peptides, liposomes, polymeric mixtures, or colloids)
- formulations (e.g., emulsions, suspensions, or nanoparticles)
- routes of delivery (e.g., locally acting drugs like inhalers, topicals, or ophthalmics)
- dosage forms (e.g., transdermal patches, extended-release injectables, or implants)
- drug-device combinations (e.g., metered-dose inhalers, auto-injectors, or nasal sprays)
Examples of drug delivery systems include inhalers, transdermal patches, long-acting injectables, liposomal injectables, and topical creams. These systems often act locally, making standard blood-level pharmacokinetic measurements inadequate. Factors such as manufacturing sensitivities, device interactions, and microstructure can significantly affect their performance.
Major Challenges in Proving Equivalence:
- Complexities in estimating absorption: Measuring absorption for locally acting products, such as inhaled or topical drugs, poses difficulties because systemic pharmacokinetics may not accurately indicate local delivery or effectiveness, resulting in challenges in identifying differences.
- Advanced characterization requirement: is necessary beyond standard PK, requiring extensive in vitro tests such as particle size distribution, dissolution profiles, and microstructure analysis to demonstrate sameness in critical quality attributes.
- Higher Variability and sensitivity: Complex products exhibit high variability and sensitivity due to potential manufacturing variations, excipient interactions, or device inconsistencies, complicating the demonstration of equivalence.
- Alternative study designs: are necessary as traditional crossover pharmacokinetic (PK) studies may be inadequate. Potential options include pharmacodynamic (PD) studies, comparative clinical endpoint studies, in vitro-in vivo correlations (IVIVC), virtual BE modeling, or combined approaches.
- Regulatory uncertainty: Regulatory pathways can differ per product, necessitating early engagement with the FDA and adaptive product-specific guidance’s.
Higher Regulatory Expectations:
Regulatory bodies such as the FDA advocate for science-based, systematic methodologies for complex generics. This includes demonstrating pharmaceutical equivalence (identical active ingredient, strength, and dosage form) along with bioequivalence, employing the most accurate, sensitive, and reproducible methods as outlined in 21 CFR 320.24.
To achieve therapeutic equivalence for generic drugs, it is essential to utilize the most accurate, sensitive, and reproducible methods as outlined in 21 CFR 320.24. This includes conducting
Figure 1: Conducting Therapeutic Equivalence
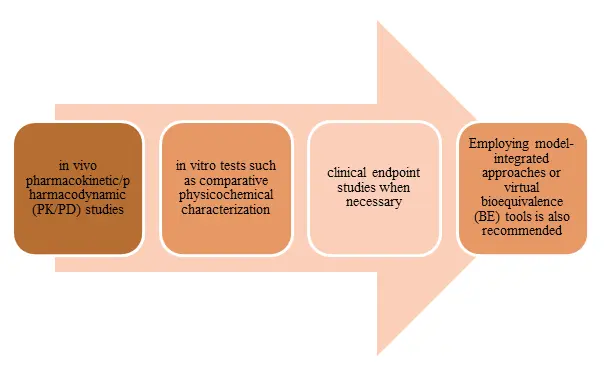
Adherence to product-specific guidances (PSGs) is critical, as the FDA provides tailored recommendations for many complex products, which are updated quarterly and may include sections dedicated to forthcoming PSGs. Engaging in early interactions through pre-submission meetings is encouraged to clarify requirements, particularly for uncertain regulatory pathways. The overarching goal is to ensure that the generic product is interchangeable with the reference listed drug (RLD) regarding safety, efficacy, and quality.
Who should read this?
This overview is particularly relevant for:
- Regulatory affairs professionals navigating ANDA submissions and FDA interactions
- Quality assurance (QA) teams ensuring compliance with characterization and stability requirements
- R&D and formulation scientists tackling development hurdles for complex dosage forms
Zenovel is dedicated to providing comprehensive support for the development of complex generics, which includes services such as bioequivalence study design, execution, advanced in vitro/in vivo characterization, regulatory strategy, and consultation aimed at meeting rigorous FDA and EMA standards. Our expertise is particularly valuable for those encountering obstacles with locally acting products, drug-device combinations, or innovative bioequivalence strategies, enabling clients to expedite the approval and market introduction processes. By linking innovation with accessibility, Zenovel aims to harness the significant potential of complex generics to lower healthcare costs and make these solutions a reality.