
Introduction to Generic Inhalation Product Development
Inhalation products pose significant challenges in generic drug development due to the complexity of formulations and device technology, making bioequivalence more difficult to establish compared to conventional oral dosage forms. Examples include Albuterol MDIs (metered dose inhalers), DPIs Dry Powder Inhalers, and nebulized therapies, which encounter various scientific, regulatory, and manufacturing obstacles in their development pathways.
Generic developers face significant challenges in the growing market for inhaled products due to a high failure rate, with estimates suggesting that 70% of pharmacokinetic studies for generic inhalers do not demonstrate bioequivalence. Recognizing these obstacles is essential for finding solutions.
👉 Accelerate Generic Inhaler Development with Expert Guidance
Navigate formulation, device, and regulatory complexities with support from inhalation development specialists.
Challenge 1: The Bioequivalence Puzzle
Why PK Studies Fail
The main challenge in generic inhaler development is proving bioequivalence (BE) to the reference product. Unlike oral medications, where systemic exposure is linked to therapeutic outcomes, inhalers target the lungs locally. This creates a paradox: while regulators demand systemic pharmacokinetic (PK) data to evaluate BE, systemic levels may not accurately represent lung deposition patterns.
The failure rate of PK studies for generic orally inhaled products (OIPs) is notably high, with approximately 70% not achieving the 80–125% bioequivalence standards. DPIs tend to be more challenging than MDIs, and combination products with multiple active ingredients further increase the difficulties faced.
👉 Struggling with Bioequivalence Failures?
Improve PK study success rates with optimized formulation strategies and regulatory-focused development support.
The IVIVC Gap
A reliable in vitro-in vivo correlation (IVIVC) for inhalation products is lacking, hindering developers’ ability to predict how formulation changes affect in vivo performance based on in vitro data. The common method of adjusting fine particle dose due to unsuccessful pharmacokinetic results is overly simplistic and often ineffective.
In vitro data is often not collected concurrently with PK studies, leading to potential mismatches in comparisons due to changes in OIPs over time, such as reductions in fine particles and lung dose when testing batches months apart.
👉 Bridge the IVIVC Gap with Scientific Expertise
Enhance in vitro and in vivo correlation using advanced analytical and dissolution methodologies.
Challenge 2: The Formulation-Device Interplay
A Combination Product Reality
Inhalation products are defined as combination products, meaning the formulation is intrinsically linked to the device. Specifically, DPIs are governed by 21 CFR 3.2(e) as combination products and must adhere to current Good Manufacturing Practices (cGMP) for both pharmaceuticals and devices, presenting a regulatory challenge for companies used to traditional oral solid formulations.
Device Platform Decisions

For emerging companies, device platform selection is a critical early decision, affecting nearly all subsequent development activities by determining the choice between DPIs, MDIs, or nebulizers.
Each platform has unique strengths and weaknesses that should be assessed in relation to the target product profile, expected dose range, and characteristics of the patient population.
👉 Select the Right Inhalation Device Platform
Get expert support in choosing between DPI, MDI, and nebulizer technologies for your product goals.
The Dose Dilemma
Inhalation products require reformulation or device modifications for dose escalation, contrasting with oral products that rely on additional tablets. This necessitates careful planning of early clinical studies to obtain adequate dose-ranging data while minimizing formulation changes.
Early-stage companies encounter challenges in generating dose-defining data due to the inadequacy of traditional animal pharmacology studies in predicting human pulmonary delivery, attributed to anatomical and physiological differences. Additionally, the high costs and complexity of early human studies with inhalation products can result in delays in obtaining essential data, often after substantial investments have been made.
👉 Simplify Dose Escalation Challenges
Design efficient dose-ranging studies while minimizing costly formulation modifications.
Challenge 3: Dissolution and Lung Biology Complexity
- The iBCS Framework
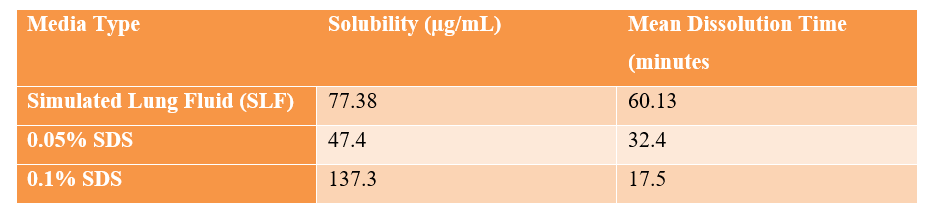
Recent advances include the Inhalation Biopharmaceutical Classification System (iBCS), published in 2023, which aims to establish a bioequivalence framework for inhaled products. However, significant shortcomings in the methodologies for measuring inhaled formulation solubility and dissolution were identified. Research indicates that the dissolution behaviour in bio-relevant media is more complex than previously understood, with solubility for budesonide, a model iBCS Class II poorly soluble drug, varying significantly across lung lining fluid simulants.
| Media Type | Solubility (µg/mL) | Mean Dissolution Time (minutes |
| Simulated Lung Fluid (SLF) | 77.38 | 60.13 |
| 0.05% SDS | 47.4 | 32.4 |
| 0.1% SDS | 137.3 | 17.5 |
The improved solubility and unrushed dissolution in lipid-based media emphasize the significance of multilamellar vesicle formation in regulating drug release, a complexity overlooked by traditional testing methods.
👉 Advance Dissolution & PBPK Understanding
Leverage modern inhalation modeling and bio-relevant testing approaches for better product predictability.
- PBPK Modeling Limitations
Integrating dissolution data with physiologically-based pharmacokinetic (PBPK) modeling poses challenges, notably for budesonide. Neglecting to consider lung retentive mechanisms, such as reversible esterification, has resulted in substantial overestimations of crucial pharmacokinetic parameters.

These findings highlight the necessity for enhanced parameterization and better integration of experimental data with PBPK modeling for inhaled products.
Challenge 4: Manufacturing and Scale-Up Complexities
Early-stage companies frequently underestimate the material requirements for regulatory-enabling studies, with initial feasibility needing about 50 g of API. Progressing to GLP inhalation toxicology studies, however, requires 0.5–2.0 kg, which poses significant scaling and financial challenges.
- Process Validation Expectations

The EMA’s updated guideline on inhalation products (February 2026) sets higher standards for manufacturing data, introducing key requirements.
👉 Prepare for Global Regulatory Expectations
Ensure your inhalation product meets evolving USFDA and EMA validation requirements.
These requirements demand careful pre-planning to avoid major objections during regulatory review.
- Scalable Technology Commitment
Early commitment to scalable technologies in laboratory-scale processes can prevent the need for expensive redesigns later, particularly in specialized manufacturing for inhalation products like spray drying, jet milling, and coarse lactose blending.
👉 Scale Your Inhalation Product with Confidence
Avoid costly redevelopment by implementing scalable manufacturing technologies from the start.
Challenge 5: Regulatory Divergence Between Markets
- US vs. EU Expectations

Inhalation product developers face **divergent regulatory requirements** across major markets :
| Parameter | USFDA | EU EMA |
| Process Validation | PPQ protocols must be adequately established prior to commercialization to ensure compliance and readiness for submission. | Full commercial scale validation data is a requirement in the Marketing Authorization Application (MAA). |
| BE Approach | Product-specific guidance indicates that pharmacokinetic (PK) studies are required for the evaluation of products. | Stepwise approach with increasing data is typically required for pharmacokinetics (PK). |
| DPI Classification | Combination product (drug + device) | Specialized pharmaceutical form |
EU requirements for pharmaceutical development and quality control can significantly differ from FDA expectations, necessitating developers seeking global approvals to create data packages that meet both authorities’ standards.
EMA’s February 2026 update to inhalation product guidelines includes new requirements: characterization of spray pattern and plume geometry, user acceptance testing for re-priming instructions, expanded robustness testing with drop impact evaluation, a section on therapeutic equivalence parameters, and lifecycle management identifying changes requiring Type II variations.
👉 Navigate Global Inhalation Regulations Efficiently
Develop harmonized data packages aligned with both FDA and EMA expectations.
Challenge 6: Batch-to-Batch Variability
- The Mismatched Batch Problem
For example: If we do a case study on tiotropium DPI, which highlights the challenges of batch-to-batch variability in bioequivalence studies, where initial PK studies failed to demonstrate bioequivalence for Cmax despite AUC being acceptable. Reanalysis with realistic impactor testing showed that in vitro ratios matched PK data, indicating previous selection of mismatched biobatches. Ultimately, equitable comparisons using this method in two additional PK studies confirmed bioequivalence with strong IVIVCs.
The REF Batch Challenge highlights that variations in the performance of reference products over time can complicate comparisons, particularly when new REF batches differ from those used in initial pharmacokinetic studies. This understanding is crucial, as it prevents developers from repeatedly encountering failed studies due to lack of insight into each product’s performance distribution.
👉 Reduce Bioequivalence Study Risks
Identify batch variability challenges early to improve study consistency and avoid repeat failures.
Challenge 7: Realistic Testing Methods
- Standard compendial cascade impactor testing
Standard compendial cascade impactor testing is crucial for quality control but often does not accurately predict in vivo performance. The tiotropium case study shows that using realistic impactor methods, which include physiological mouth-throat models and patient-specific inspiratory profiles, yields better IVIVCs than traditional methods. Key aspects of realistic testing are the use of OPC mouth-throat models, simulated inspiratory profiles, and whole lung dose estimation. Additionally, current dissolution methodologies for inhaled products are inadequate; modified Andersen cascade impactors show potential, yet challenges remain in solubility assessment and dissolution studies.

Zenovel specializes in inhalation product development, leveraging over a years of regulatory and technical experience to assist clients and nationally globally. Our team aids sponsors in navigating the intricacies of generic inhaler development, encompassing formulation optimization and regulatory submission.
Zenovel offers extensive services for the development of generic inhalation products, leveraging strong regulatory knowledge alongside technical expertise in formulation, analytical methods, and manufacturing.
Ready to Advance Your Inhalation Product Development? Zenovel offers expertise in inhalation product development to help accelerate regulatory approval processes. Contact us for further discussion.
👉 Upgrade to Realistic Inhalation Testing Approaches
Adopt patient-relevant testing methodologies that better predict in vivo performance.
References:
Guideline on the pharmaceutical quality of inhalation and nasal medicinal products II 14 July 2025 EMEA/CHMP/QWP/49313/2005 rev. 1 Committee for Medicinal Products for Human Use (CHMP)
Berg C, Watts A, “Charting the Course for Pulmonary Drug Development”. ONdrugDelivery, Issue 180 (Nov 2025), pp 10–14.
Current Challenges for the Development of Inhaled Medicines
Paramanandana, A. (Author). 1 Jun 2025
Nashwa El-Gendy, Craig M. Bertha, Mohammed Abd El-Shafy, et al. Scientific and regulatory activities initiated by the U.S. food and drug administration to foster approvals of generic dry powder inhalers: Quality perspective. Advanced Drug Delivery Reviews. Vol 18 2022.
Sandell D. Bioequivalence assessment of pharmaceutical aerosol products through IVIVC. Advanced Drug Delivery Reviews. https://doi.org/10.1016/j.addr.2021.113895
Gobetti C. Bioequivalence of Two Tiotropium Dry Powder Inhalers and the Utility of Realistic Impactor Testing. Journal of Aerosal Medicine and Pulmonary Drug Delivery
👉 Ready to Advance Your Generic Inhalation Product?
Partner with Zenovel for expert support in formulation development, analytical testing, manufacturing, and regulatory strategy.